


377-2 Ohno-Higashi, Osaka-Sayama, Osaka 589-8511, Japan
Tel +81-72-366-0221
Fax +81-72-366-0245
E-mail seika2@med.kindai.ac.jp
Outline of our study

Our aims
In Japan, one in two people will develop cancers, and one in four people will die of these malignant diseases. Because of the expansion of the aging population, cancer incidence and mortality are expected to increase further. Thus, further understanding of cancer biology and applying our knowledge to improve early diagnosis and treatment efficacy is becoming increasingly critical. In spite of significant progress in medical science and cancer therapy, major challenges remain, which include cancers with difficulties in early diagnosis, therapeutic resistance, recurrence, and metastasis.
Cancer development, or tumorigenesis, is driven by dynamic interactions between environmental factors and genetic diversities/abnormalities. Technological advances, such as next generation sequencing, have enormously expanded our understanding of cancer biology. However, key questions persist: How do normal cells, precancerous and cancer cells respond intrinsic and extrinsic selection pressures during malignant transformation? How is cell fate determined, and what govens these selection processes? Recent evidence suggests that cancer cells adapt to their environment and evade cancer surveillance mechanisms through epigenetic reprogramming at the very early stages of malignant transformation, enabling their survival and sustained proliferation. To tackle these issues, we aim to explore the pathophysiology of cancer through the lenses of environmental response and epigenetic regulation, ultimately identifying novel therapeutic targets and diagnostic markers for prevention and treatment.
Epigenetics refers to “stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”, encompassing the modifications of DNA binding proteins (e.g. histones), DNA, RNA, and noncoding RNAs. Growing evidence strongly highlights the role of epigenetics in shaping biological properties of cancer cells from their very early to later stages of proliferation. Thus, in order to develop an innovative strategy for cancer prevention and treatment, we need deeper understandings of the regulatory mechanisms governing cellular responses to intrinsic and extrinsic changes in vivo. To address these questions, we mainly employ genetically modified mouse models and cultured cells to study their phenotypic alterations through epigenomic, cell biological, biochemical approaches and imaging approaches. Furthermore, we are expanding our research into aging biology to uncover potential routes for cancer prevention.
Outlines of our past research
1. Apoptosis regulation
We have studied apoptosis as a phenotype of environmental responses. Deregulated apoptosis is a hallmark of cancer. In fact, deregulation of apoptosis regulators is closely associated with multiple processes of tumorigenesis. We are focusing on the genes involved in nuclear signaling and apoptosis regulation, and examine the underlying molecular mechanisms with special emphasis on the TP53 signaling pathway.
Generation and characterization of Smac/Diablo-deficient mice. Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X, Mak TW. Mol Cell Biol. 22:3509-3517 (2002)
Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. Okada H*, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, Ciofani M, Rottapel R, Zuniga-Pflucker JC, Mak TW. J Exp Med. 199:399-410 (2004) (*Corresponding author)
Pathways of apoptotic and non-apoptotic death in tumour cells. Okada H, Mak TW. Nat Rev Cancer 4(8): 592-603 (2004)
Bat3 deficiency accelerates the degradation of Hsp70-2/Hspa2 during spermatogenesis. Sasaki T, Marcon E, McQuire T, Arai Y, Moens PB, Okada H. J Cell Biol. 182:449-458 (2008)
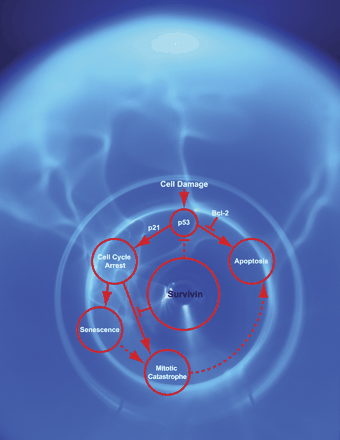
J Exp Med. Vol. 199, 3. Cover art by Graham Hutcheson
2. DNA damage response
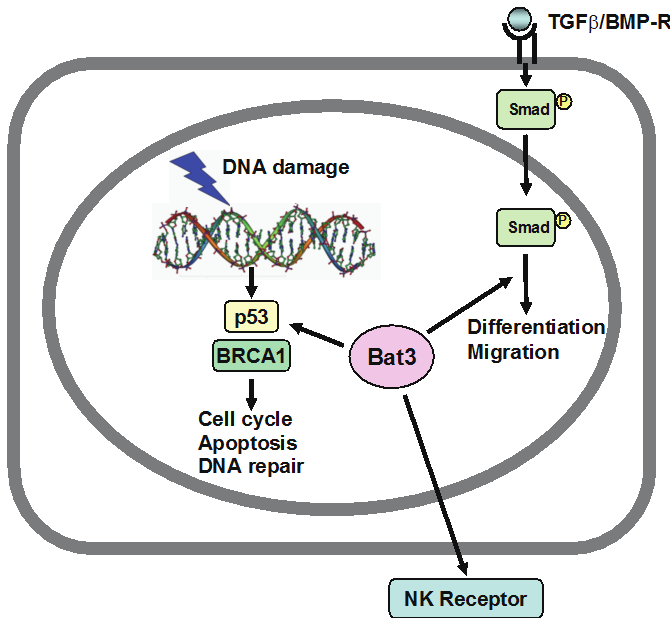
As a response to extrinsic environmental signals, we focus on DNA damage response. Deregulation of DNA damage response and repair mechanisms lead to genomic instability, malignant transformation and heterogeneity of cancer, thus contributing to malignancy grade and aggravating therapeutic response. DNA damage response also has associated with regulation of epigenetics. We are focusing on the regulatory mechanisms of DNA double-strand break repair.
HLA-B-associated transcript 3 (Bat3)/Scythe is essential for p300-mediated acetylation of p53. Sasaki T, Gan EC, Wakeham A, Kornbluth S, Mak TW, Okada H. Genes Dev. 21:848-861 (2007)
Attenuated DNA damage repair delays therapy-related myeloid neoplasms in a mouse model. Tong KI, Ota K, Komuro A, Ueda T, Ito A, Koch AC, Okada H. Cell death & Disease Oct 6;7(10):e2401 (2016 )
Ascorbate sensitizes human osteosarcoma cells to the cytostatic effects of cisplatin. Oka N, Komuro A, Amano H, Dash S, Honda M, Ota K, Nishimura S, Ueda T, Akagi M, Okada H. Pharmacology Research & Perspectives (2020 Aug 8; 8(4)e00632)
3. Metabolism & Epigenetics
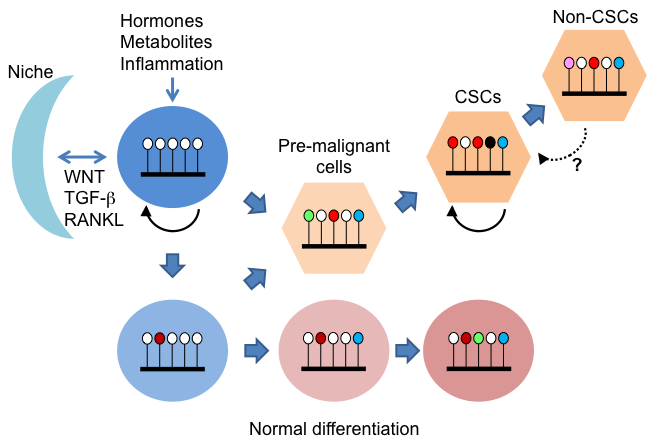
In addition to genetic alterations, long- and short-term gene expression patterns established by epigenetics in response to environmental signals make a significant contribution to malignant transformation. However, the details of the epigenetics-mediated regulation of tumorigenesis are still unclear, especially in vivo. We are studying their roles in cancer development and metabolism by focusing on the regulatory mechanisms of histone methylation and chromatin remodeling.
Histone demethylase Jmjd2b functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son D-O, Wakeham A, Miyagishi M, Mak TW, Okada H. PLoS ONE 6:e17830 (2011)
The H3K27 demethylase, Utx, regulates adipogenesis in a differentiation stage-dependent manner. Ota K, Tong KI, Goto K, Tomida S, Komuro A, Wang Z, Nishio K, Okada H. PLOS ONE 12(3): e0173713 (2017)
JMJD2B/KDM4B inactivation in adipose tissues accelerates obesity and systemic metabolic abnormalities. Kang C, Saso K, Ota K, Kawazu M, Ueda T, Okada H. Genes Cells (online 2 August, 2018)
High Fat Diet Triggers a Reduction in Body Fat Mass in Female Mice Deficient for Utx demethylase. Ota K, Komuro A, Amano H, Kanai A, Ge K, Ueda T, Okada H. Scientific Reports (11 July, 2019)
KDM4B promotes acute myeloid leukemia associated with AML-ETO by regulating chromatin accessibility. Ueda T*, Kanai A, Komuro A, Amano H, Ota K, Honda M, Kawazu M, Okada H*. (*co-correspondence) FASEB BioAdvances 29 August 2021.
MYC/glutamine dependency is a therapeutic vulnerability in pancreatic cancer with deoxycytidine kinase inactivation-induced gemcitabine resistance. Dash S, Ueda T, Komuro A, Amano H, Honda M, Kawazu M, Okada H. Mol Cancer Res.(2023) 21 (5): 444–457 (online publication Feb 9, 2023)
Deoxycytidine Kinase Inactivation Enhances Gemcitabine Resistance and Sensitizes Mitochondrial Metabolism Interference in Pancreatic Cancer. Dash S, Ueda T, Komuro A, Honda M, Sugisawa R, Okada H*. Cell Death & Disease (2024) 15:131
Vgll2 as an integrative regulator of mitochondrial function and contractility specific to skeletal muscle. Honda M, Inoue R, Nishiyama K, Ueda T, Komuro A, Amano H, Sugisawa R, Dash S, Shirakawa J, Okada H. J Cell Physiol. (2024) e31436 109940; DOI:10.1002/jcp.31436